

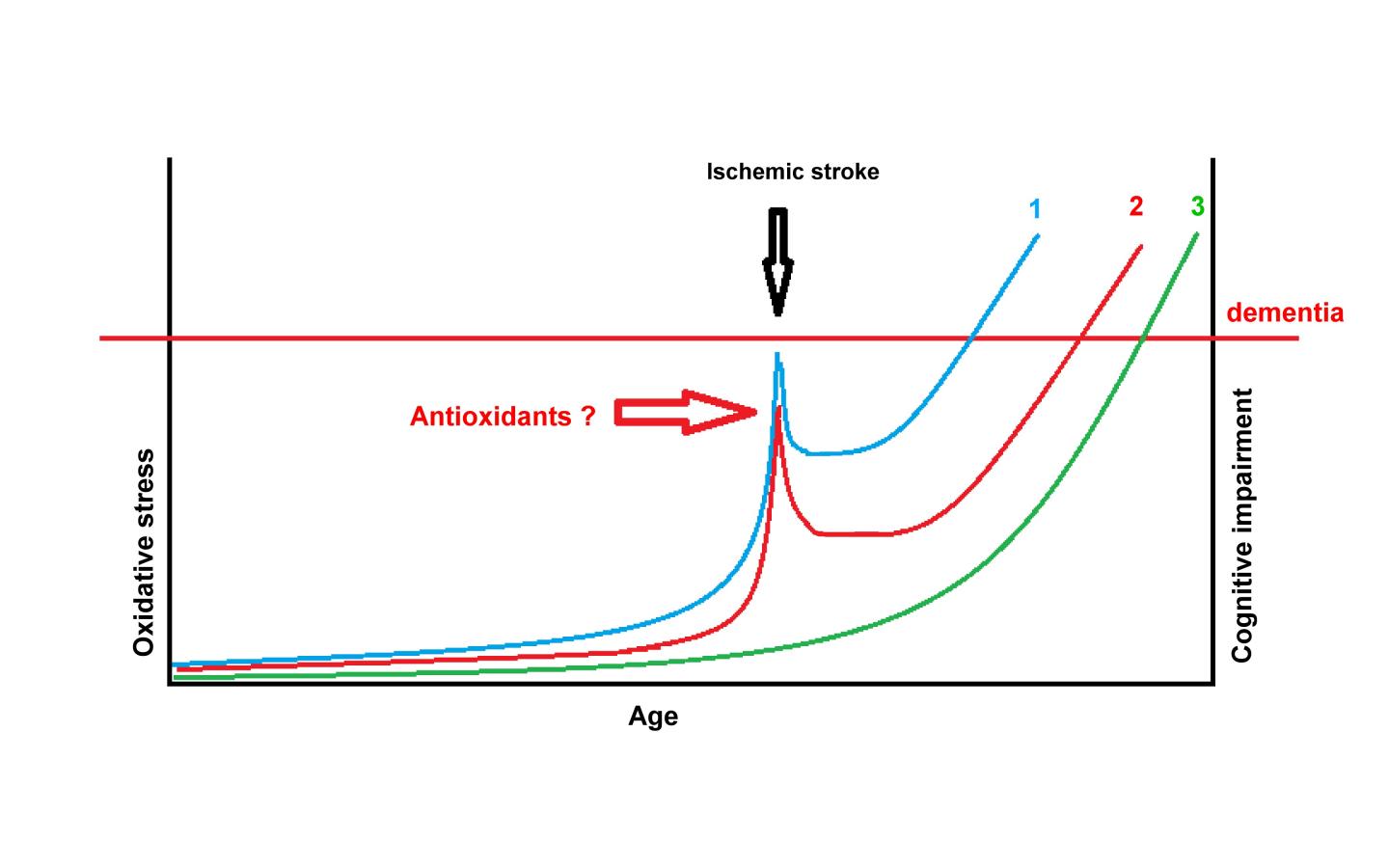
IMAGE: With normal ageing oxidative stress increases and is involved in causing degenerative diseases like Alzheimer’s disease. An ischemic stroke significantly increases oxidative stress in the brain, and this may…
view more
Credit: © Bentham Science Publishers
Currently we are facing a dementia epidemic, estimations showing that by 2050 approximately 131 million people will be affected. Every 7 seconds a patient is diagnosed worldwide. Because the common forms of dementia occur in the elderly, delaying the onset or worsening of the cognitive impairment could translate into a significant reduction of the incidence of the disease. Estimations have shown that of the huge number of cases expected by 2050, roughly 23 million could be avoided if the onset of the disease could be delayed by 2 years. Despite the ambition to identify a disease modifying therapy or a cure for dementia by 2025 set by the G8 dementia summit in 2013, the findings so far are not very encouraging.
To date there is growing evidence of the association of vascular risk factors like hypertension, high cholesterol levels or diabetes mellitus with cognitive impairment and Alzheimer’s disease. Unfortunately, simply managing these risk factors had little effect in reducing the incidence of dementia. These factors, however, strongly increase the risk of a patient to suffer an ischemic stroke and incident stroke approximately doubles the risk of dementia. From the study of Saver published in 2006 we know that “each hour in which treatment fails to occur the brain loses as many neurons as it does in 3.6 years of normal aging”.
These neuronal losses occur through ischemic necrosis in the core of the infarction, but may be prolonged up to 2 weeks after the ischemic insult in the penumbral area surrounding the ischemic core through another type of cell loss, namely apoptosis. In initiating apoptosis oxidative species have a major role. Several authors have shown consistent increases in oxidative stress after an ischemic stroke. As the authors pointed out in a previous study, oxidative stress increases mainly after cardioembolic stroke, followed by lacunar stroke, with a less prolonged burst of generation of oxidative species following thrombotic stroke.
There is a considerable overlap between the oxidative stress-induced pathogenesis in ischemic stroke and Alzheimer’s disease including mitochondrial dysfunction (the mitochondria being the main generators of energy in the cells), calcium overload of the cells, activation of different destructive enzymes by the excess intracellular calcium, aberrant gene transcription and expression, induction of autophagy (a process by which cells degrade their own cytoplasmic proteins and organelles) and activation of inflammatory responses.
Despite promising results of antioxidant molecules in animal models of ischemic stroke, human clinical trials were disappointing possibly due to late administration and incorrect selection of patients. However, in a study published in 2019 edaravone (an antioxidant molecule) given within 48 hours after endovascular revascularization in acute ischemic stroke was associated with greater functional independence at hospital discharge, lower in-hospital mortality and reduced intracranial hemorrhage after admission in a study which enrolled over 10,000 patients. More recently in a report presented at the International Stroke Conference 2020, nerinetide or NA1, a molecule which reduces endogenous nitric oxide (also an oxidative species) generated inside the cell during ischemia, improved the outcome of ischemic stroke patients who underwent endovascular thrombectomy. Unfortunately, NA1 interacted with alteplase, limiting its efficiency in patients who were also thrombolysed.
Antioxidants have been evaluated also in degenerative diseases, Alzheimer’s disease included, with promising results in animal models but inconclusive results in clinical trials. Therapeutic strategies are hampered by the dual role of oxidative species in the organism. On one hand, increased ROS production contributes to age-related chronic conditions and on the other, oxidant species function as signaling molecules in pathways that are critical for cell survival. However, based on the compelling evidence of the implication of oxidative stress in AD pathogenesis and of the pivotal role of mitochondria, molecules acting as mitochondria-targeted antioxidants show promise in animal models of neurodegenerative diseases, improve mitochondrial function after coronary ischemia/reperfusion in rats, and some have already been developed into drugs used in clinical trials in type 2 diabetic patients.
In view of the implication of oxidative stress in the genesis of AD pathology, the authors hypothesize that with aging, in the presence of well-established vascular risk factors, and possibly with a genetic contribution, AD pathology develops slowly without clinically overt cognitive impairment. However, after a stroke there is a sudden burst in oxidative stress which accelerates the pathogenesis of dementia and leads to clinically obvious cognitive impairment. If this hypothesis would be proven the reason for reaching antioxidant treatment in acute ischemic stroke would be reinforced. Further studies in this direction with long follow-up periods would be needed. Nonetheless, in view of the high incidence and prevalence of the disease, the results could be rewarding.
###
Keywords: Alzheimer’s disease, cerebrovascular disease, dementia, pathogenesis, neurovascular unit, oxidative stress, antioxidant treatment.
For further information, please visit: https:/
TDnews














{kind=link}